
Late-onset hepatoerythropoietic porphyria presenting with facial deformities and erythrodontia
Keywords:
Hepatoerythropoietic porphyria, facial deformities, erythrodontia, late-onsetAbstract
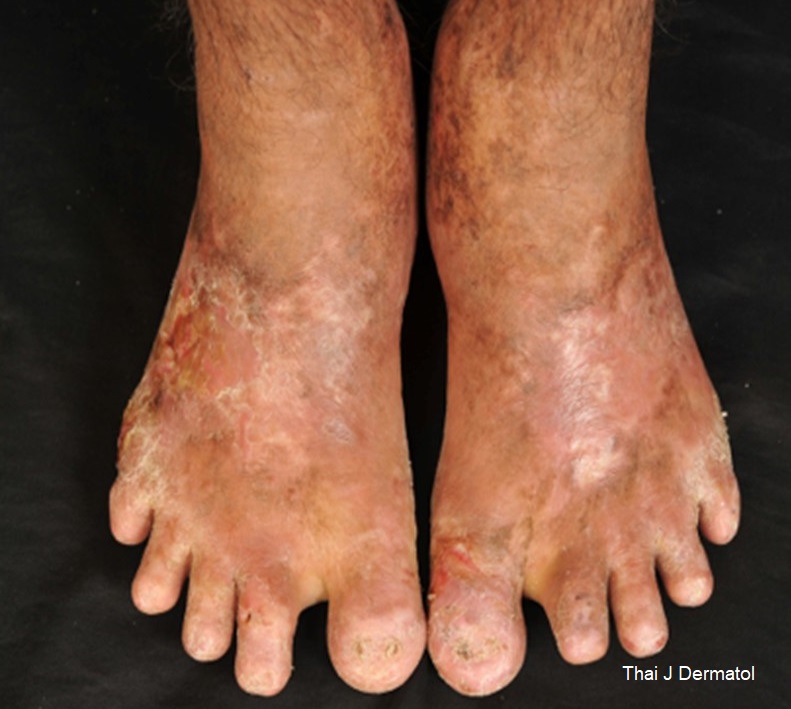
Hepatoerythropoietic porphyria (HEP) is a nonacute porphyria inherited as an autosomal recessive disorder due to either a homozygous or compound heterozygous deficiency of uroporphyrinogen decarboxylase (UROD). The incidence of HEP is extremely rare, with fewer than 40 cases reported to date. The typical onset is in early childhood, and it usually presents with severe photosensitivity, mutilating scarring, and hypertrichosis. We report a 48-year-old man with teenage onset (15 years of age) of HEP.
References
2. Peerapittayamongkol C, Srisawat C, Junnu S, Kanyok S, Soiampornkul R, Poonsawas I, Neungton N. Porphyrias diagnosed types by HPLC analysis: 10 years of Siriraj experience. Siriraj Med J 2007;59:356-60.
3. Deacon AC, Elder GH. ACP Best Practice No 165: front line tests for the investigation of suspected porphyria. J Clin Pathol 2001; 54: 500-7.
4. Moran-Jimenez MJ, Ged C, Romana M, et al. Uroporphyrinogen decarboxylase: complete human gene sequence and molecular study of three families with hepatoerythropoietic porphyria. Am J Hum Genet 1996; 58: 712-21.
5. Balwani M, Desnick RJ. The porphyrias: advances in diagnosis and treatment. Blood 2012; 120: 4496-504.
6. Cantatore-Francis JL, Cohen-Pfeffer J, Balwani M, et al. Hepatoerythropoietic porphyria misdiagnosed as child abuse: cutaneous, arthritic, and hematologic manifestations in siblings with a novel UROD mutation. Arch Dermatol 2010; 146: 529-33.
7. Karim Z, Lyoumi S, Nicolas G, Deybach JC, Gouya L, Puy H. Porphyrias: A 2015 update. Clin Res Hepatol Gastroenterol 2015; 39: 412-25.
8. Phillips JD, Whitby FG, Stadtmueller BM, Edwards CQ, Hill CP, Kushner JP. Two novel uroporphyrinogen decarboxylase (URO-D) mutations causing hepatoerythropoietic porphyria (HEP). Transl Res 2007; 149: 85-91.
9.Ramanujam VM, Anderson KE. Porphyria Diagnostics-Part 1: A Brief Overview of the Porphyrias. Curr Protoc Hum Genet 2015; 86: 17.20.1-26.
10. Remenyik E, Lecha M, Badenas C, et al. Childhood-onset mild cutaneous porphyria with compound heterozygotic mutations in the uroporphyrinogen decarboxylase gene. Clin Exp Dermatol 2008; 33: 602-5.
11. Armstrong DK, Sharpe PC, Chambers CR, Whatley SD, Roberts AG, Elder GH. Hepatoerythropoietic porphyria: a missense mutation in the UROD gene is associated with mild disease and an unusual porphyrin excretion pattern. Br J Dermatol 2004; 151: 920-3.
12. Horina JH, Wolf P. Epoetin for severe anemia in hepatoerythropoietic porphyria. N Engl J Med 2000; 342: 1294-5.
Downloads
Published
How to Cite
Issue
Section
License
เนื้อหาและข้อมูลในบทความที่ลงตีพิมพ์ในวารสารโรคผิวหนัง ถือเป็นข้อคิดเห็นและความรับผิดชอบของผู้เขียนบทความโดยตรงซึ่งกองบรรณาธิการวารสาร ไม่จำเป็นต้องเห็นด้วย หรือร่วมรับผิดชอบใดๆ
บทความ ข้อมูล เนื้อหา รูปภาพ ฯลฯ ที่ได้รับการตีพิมพ์ในวารสารโรคผิวหนัง ถือเป็นลิขสิทธิ์ของวารสารฯ หากบุคคลหรือหน่วยงานใดต้องการนำทั้งหมดหรือส่วนหนึ่งส่วนใดไปเผยแพร่ต่อหรือเพื่อกระทำการใดๆ จะต้องได้รับอนุญาตเป็นลายลักอักษรจากบรรณาธิการวารสารโรคผิวหนังก่อนเท่านั้น

